
Fast Ant Colony Clustering for Functional Module Detection Algorithm in PPI Networks
-
摘要:
针对蚁群聚类在蛋白质相互作用(protein-protein interaction,PPI)网络中进行功能模块检测问题上时间性能的不足,提出一种快速的基于蚁群聚类的PPI网络功能模块检测(fast ant colony clustering for functional module detection, FACC-FMD)方法. 该算法计算每个蛋白质与核心组蛋白质的相似度,根据拾起放下模型进行聚类,得到的初始聚类结果中功能模块之间相似度很小,省去了原始蚁群聚类算法中的合并和过滤操作,缩短了求解时间. 同时该算法根据蛋白质的关键性对蚁群聚类中的拾起放下操作做了更严格的约束,以减少拾起放下的次数,加速了聚类的过程. 在多个PPI网络上的实验表明:与原始蚁群聚类方法相比,FACC-FMD大幅度提高了时间性能,同时取得了良好的检测质量,而且与近年来的一些经典算法相比在多项性能指标上也具有一定的优势.
Abstract:The time performance of ant colony clustering seriously restricts its application for functional module. A fast ant colony clustering for functional module detection (FACC-FMD) algorithm, which considerably speeded up the original ACC-FMD algorithm was developed. The similarity between each protein and core protein group was computed by the FACC-FMD, then clustered by the pick-up and drop-down model. The similarity between the functional modules by clustering was small. Thus FACC-FMD eliminated the need for the merge operation and filter operation in ant colony cluster, and shorten the running time. At the same time, the essential of protein was computed and was used to constraint the times of pick-up and drop-down. Experiments on multiple PPI networks show that the FACC-FMD algorithm can greatly improve the time performance of ant colony clustering for functional module detection with satisfactory quality. Moreover, compared with classical algorithms in recent years, the FACC-FMD also has advantages in performance indicators.
-
蛋白质是所有生命的物质基础,也是一切生命活动的体现者. 蛋白质在参与生命活动时,很少以独立个体的方式存在,而是以通过相互作用构成大分子复合物的形式完成对应的生物学功能. 在一个生命有机体内,所有蛋白质之间的相互作用组成的生物分子关系网络叫作蛋白质相互作用(protein-protein interaction,PPI)网络. 不同的时间和空间阶段通过相互作用共同参与某一特定分子进程的蛋白质集合称为功能模块. 对PPI网络进行研究与分析的一个主要目的是检测出其中的功能模块,而聚类是功能模块检测的常用方法之一. 从PPI网络数据中检测功能模块是后基因时代蛋白质组学研究的重要内容,它有助于理解蛋白质之间的相互作用以及各种生物学过程,能够揭示疾病的发生机制,为新药研发提供重要的理论基础 [ 1] .
近10年间,大规模PPI网络数据的获得,使得以数据挖掘、机器学习为基础的计算方法迅速兴起. 依据所采用的计算模型和机理,可以将其划分为基于传统图理论的方法和基于非传统图理论的方法两大类 [ 2] . 其中,基于传统图理论的检测方法依据PPI网络的拓扑结构信息完成聚类,主要有基于密度的检测方法、基于层次的检测方法、基于划分的检测方法等 [ 3] . 例如,文献[4]提出了基于密度的检测(molecular complex detection,MCODE)方法. 该方法首先选取局部邻域密度最大的节点作为初始的功能模块,然后向外扩张该节点形成最终的功能模块. MCODE能够有效地检测出密度大的功能模块,但在稀疏的PPI网络中效果不佳. 文献[5]提出了基于层次的检测方法JERARCA,该方法先计算PPI网络的距离矩阵,再将PPI网络的距离矩阵转换成为层次结构的树,然后根据模块内和模块间节点的连接分布进行最优层次划分得到功能模块. JERARCA方法在聚类过程中某个节点的层次分类错误,将导致其下层节点的分类不正确,因此该方法对噪声数据非常敏感. 基于非传统图理论的检测方法是将其他机理融合于图聚类过程,主要有基于流模拟的检测方法、基于核心-附属关系的检测方法、基于群智能的检测方法等. 例如,文献[6]提出了一种基于流模拟的马尔可夫聚类(Markov clustering,MCL)检测方法,该方法重复模拟流在PPI网络中的扩展和收缩行为,将PPI网络划分为许多稠密子图作为最终的检测结果. MCL能够适应网络的变化,在一定程度上克服噪声数据的影响,具有很强的鲁棒性. 依据对实验所确定的蛋白质复合物的生物信息学分析,文献[7]提出了基于核心-附属关系的检测方法COACH. 该方法先抽取核心蛋白质,然后将附属蛋白质逐个分配到核心蛋白质周围,构成一个功能模块. COACH算法具有较强的功能模块识别能力,且能够检测出重叠的功能模块. 除此之外,近年来涌现了许多将群智能思想融合于图聚类过程的检测方法,该类算法通过模拟社会型生物群体间的协作行为实现功能模块的检测,展现了良好的检测质量. 文献[8]提出了改进的基于蚁群优化的PPI网络功能模块检测(new ant colony optimization functional module detection,NACO-FMD)方法,该方法结合PPI网络拓扑信息和基因本体(gene ontology)信息,设计PPI网络下的启发函数来指导蚁群寻优,得到了较好的检测结果. 文献[9]提出了基于多智能体进化机制的PPI网络功能模块检测方法MAE-FMD,该算法首先对PPI网络进行多智能体的解编码处理;然后通过随机游走的方式为每个智能体构建初始功能模块;最后通过3种进化算子实现种群的进化,取得了较好的结果. 文献[10]将蚁群聚类思想应用到PPI网络功能模块检测问题上,提出了基于蚁群聚类的PPI网络功能模块检测(ant colony cluster for functional module detection,ACC-FMD)方法,该算法首先选取种子节点,然后采用蚂蚁的拾起放下模型对网络中的蛋白质节点进行聚类,同时利用每代蚂蚁中的最优聚类结果和蛋白质之间的功能相似性评分更新相似性函数,将信息在不同代之间传递. 为提高检测质量,得到初始聚类结果后,ACC-FMD利用合并与过滤2个后处理操作对聚类结果进行修正,得出最终的聚类结果. 该方法与近年来一些流行的功能模块检测方法相比,具有一定的优势. 然而,该方法需要进行重复的拾起放下和大量的功能模块合并操作,导致求解时间过长.
为了克服ACC-FMD求解时间长的缺陷,本文提出一种快速的基于蚁群聚类的PPI网络功能模块检测(fast ant colony cluster for functional module detection,FACC-FMD)方法. FACC-FMD方法抽取稠密且具有高度共表达特性的子图作为核心组蛋白质,由于子图间具有较低的相似性,初始的聚类结果不需要合并,省去了大量的后处理时间. 而且该算法根据关键蛋白质的结构特性和生物特性计算蛋白质的关键性,并依据关键性高低对拾起放下操作进行了更严格的约束,从而减少了拾起放下的次数,加速聚类的过程.
1 相关工作
1.1 ACC-FMD方法
生物学家研究发现蚁群会将蚁穴中分散的蚂蚁尸体堆积成相对集中的几个大堆. 通过对蚁群清扫蚁穴行为的观察,学者们提出了蚁群聚类方法. 若将这些分散的蚂蚁尸体视为待聚类的数据对象,那么堆积而成的大堆则看作最终的聚类结果 [ 11] . 文献[10]将蚁群聚类技术运用到PPI网络的功能模块检测问题上,提出了ACC-FMD方法. 该方法将PPI网络表示为一个无向图 G=( V, E),其中 V表示蛋白质节点集合, E表示相互作用的集合. ACC-FMD算法主要包括如下4个过程.
1) 选取种子节点:种子节点是指在网络中局部密度较高的节点. ACC-FMD根据节点的聚类系数选取种子节点,将聚类系数大于阈值 ω的节点挑选出来构成种子节点集合 S,其中每个种子节点标注一个功能模块 . 对于网络 G=( V, E)中的任意节点 i,其聚类系数定义为
φ i=
式中:Neigh( i)为节点 i的直接邻居集合; n i 为集合Neigh( i)中的节点之间相互作用的个数 .
2) 聚类过程:得到种子节点集合后,蚂蚁开始遍历每个种子节点的邻域进行聚类,节点邻域定义为
N( i) =Neigh( i)∪InNeigh( i) (2)
式中InNeigh( i)表示种子节点 i的间接邻居集合 . 在遍历过程中通过拾起概率模型确定是否拾起某个节点 . 若拾起该节点,则利用放下概率模型将已拾起的节点聚到其他种子节点标注的功能模块中,然后,进行新一轮的拾起放下操作;若没有拾起,则直接将它归属到该种子节点标注的功能模块中 . 当蚂蚁遍历完所有的种子节点时,形成自身的聚类结果 . 拾起概率模型 P p( j)和放下概率模型 P d( j)分别定义为
P p( j) =
P d( j) =
式中: s( i, j)为节点 i和 j的结构相似度; k p和 k d为2个参数 . 结构相似度 s( i, j)通过归一化共同邻居定义,一般描述为
s( i, j) =
式中 τ( i)为由节点 i的直接邻居和节点 i自身构成的集合 .
3) 信息传递:ACC-FMD通过节点之间的相似度将上一代最优解的信息传递给下一代 . 该方法通过模块化密度来评价解的质量,其定义为
D=
式中: m为预测到的功能模块的数量; l h 为模块 h中所包含的节点之间存在的边数;
s( i, j) =(1 +f ij ) count s( i, j)(7)
式中: i和 j为属于同一功能模块的2个节点;count为节点 i和 j聚在同一个功能模块中的次数; f ij 为 i和 j的功能相似性评分,通过蛋白质的功能注释信息计算,定义为
f ij=
4) 后处理过程:经过一定次数的迭代后,对初始聚类结果进行合并与过滤2个后处理操作 . 合并操作是指合并2个相似度大于阈值的模块 . 模块 M x 和 M y 的相似度定义为
S( M x , M y ) =
r( i, j) =
对合并后的聚类结果过滤掉密度小于阈值的模块,形成最终的聚类结果 . 模块密度定义为
Density G =
ACC-FMD算法具有蚁群聚类思想的正反馈性、自组织性和健壮性等优点 . 然而,该算法时间性能比较差,主要归于如下2个原因:1)算法抽取种子节点,根据相似度将非种子节点聚在种子节点周围构成功能模块 . 如果2个种子节点之间相似度比较大,那么聚类所形成的2个功能模块也较为相似,在后处理过程中,需要将这2个模块合并,而合并操作需要大量的计算,影响算法的时间性能 . 2)一个蛋白质节点可能在多个种子节点的邻域内,这样在聚类过程中会出现大量重复的拾起放下操作,导致聚类时间变长 .
1.2 基因表达信息与蛋白质共表达特性
生物学研究结果表明蛋白质在生命活动中具有一系列的特性,挖掘并利用这些特性有望提升功能模块检测方法的时间性能和质量 . 生物学家在实验中观察到超过一半的酵母菌基因呈现周期性表达的现象 . 基因编码蛋白质表现出类似的模式,特别是与能量和代谢相关的基因往往表现出强大的周期性 [ 12] . 基因表达信息可以直观地描述这一现象 . 具体来说,基因表达信息是用来表示蛋白质生命活动过程中的一组数据,它记录了蛋白质在多个时刻下的基因表达值 . 基因表达值越高,说明这个蛋白质在该时刻活性越高,越具有强表达性 . 如果用函数Exp( x, t)表示蛋白质 x在某时刻 t的表达值,随着 t的变化,该函数值会表现出周期性 . 尽管每个蛋白质都具有自己的表达周期以及表达强弱,但不同的蛋白质之间具有共表达特性 . 共表达特性从基因层面上描述了蛋白质之间的关系 . 利用这一特性可以在PPI网络中抽取出一部分特殊的蛋白质,辅助完成聚类 .
本文使用皮尔逊相关系数(Pearson correlation coefficient,PCC)来度量2个蛋白质之间共表达的强弱程度 . 若蛋白质 x和 y的表达函数为Exp( x, t)和Exp( y, t),则皮尔逊相关系数的定义为
Pcc( x, y) =
式中: k为样本数,即在基因表达信息中的时刻数;
本文通过蛋白质之间的共表达特性来抽取核心组蛋白质和关键蛋白质.
功能模块一般由核心组蛋白质和附属蛋白质组成,相比之下,核心组蛋白质更能代表所处的功能模块. 从网络拓扑角度来描述,核心组蛋白质通常为小而稠密的子图 [ 13] ,核心组蛋白质之间的相互作用非常紧密. 从生物角度来描述,核心组蛋白质具有较高的共表达特性,并且被附属蛋白质包围 [ 13] .
关键蛋白质是细胞生命活动中所必需的蛋白质,Winzeler等 [ 14] 将关键蛋白质定义为:通过基因剔除式突变将其移除后会造成有关蛋白质复合物功能的丧失,并导致生物体无法生存的蛋白质. 文献[15]指出,一个蛋白质与邻近蛋白质的相互作用越多,这个蛋白质对细胞的生存越重要,且成为关键蛋白质的可能性就越高. 并且该文献也指出在这些蛋白质中,与邻居节点具有较高共表达程度的蛋白质更易成为关键蛋白质. 由于关键蛋白质在功能上的重要性,它更倾向于成为功能模块内的蛋白质.
2 FACC-FMD方法
2.1 算法思想
ACC-FMD算法的大部分时间消耗在拾起放下和合并操作上,如何有效限制拾起放下操作次数的同时,增大求得的功能模块间的差异性从而减少合并操作,就成为ACC-FMD提升效率的关键.
FACC-FMD算法结合基因表达信息,生成稠密且具有高度共表达特性的核心组蛋白质,再通过蚁群聚类机制将蛋白质归属到核心组蛋白质周围以形成功能模块. 核心组蛋白质是一个子图,由于子图之间的差异性比原蚁群聚类算法中种子节点之间的差异性大,故求得的功能模块可能不再需要进行合并操作,从而有望提高算法的时间性能. 另外,相比于非关键蛋白质,关键蛋白质对功能模块的重要性更高. 以此为依据,FACC-FMD计算每个蛋白质的关键性,利用关键性对拾起放下进行严格的约束,从而减少拾起放下次数,提高聚类的效率.
2.2 基于基因表达信息的核心组蛋白质抽取
大量的合并操作会在一定程度上影响ACC-FMD算法的时间效率. 针对这一问题,FACC-FMD算法使用核心组蛋白质替代种子节点进行蚁群聚类. 依据前面的描述,核心组蛋白质应该具有以下2个特性:第一,核心组蛋白质是小而稠密的子图;第二,核心组蛋白质具有高度的共表达特性 . 依据这2个特性,新算法首先计算每个节点的聚类系数并且与设定的阈值进行比较,将大于阈值的节点抽取出来作为潜在的核心节点集Core . 然后,将Core中的节点扩展为核心组蛋白质 . 对于Core集合中每个节点 C i ,将 C i 和它的所有邻居放入一个集合 V C,将它们之间的边放入到一个集合 E C,构成子图 G C =( V C, E C),再计算子图 G C的密度和共表达值 . G C的密度由式(11)计算得出 . G C的共表达值用子图内节点之间的平均皮尔逊相关系数描述,定义为
coExp( G C) =
式中 n为核心内的节点数,共表达值的取值范围是[ -1,1],同样地,其值越大,表示 G C的共表达程度越高 . 对于扩展得到的核心子图 G C,它的密度值和共表达值可能会小于设定的阈值 λ和 δ,这时就要递归地删除 G C中的节点,直到满足阈值的要求,形成最终的核心组蛋白质 . FACC-FMD删除关键性最低的节点,这是因为关键性高的蛋白质对生命活动更为重要,从而求得的核心组蛋白质更能体现功能模块的生物特性. 核心组蛋白质是一个稠密子图,因为子图之间的差异性较大,所以FACC-FMD可能不需要进行合并操作和过滤操作.
2.3 基于基因表达信息的蛋白质关键性计算
在ACC-FMD算法中,蛋白质节点在种子节点邻域内的次数就是其拾起放下次数. 为了克服该算法重复拾起放下次数过多的缺陷,FACC-FMD算法依据关键蛋白质的特征,计算每个蛋白质的关键性,利用关键性对拾起放下操作进行严格的约束,减少非关键蛋白质的拾起放下操作.
FACC-FMD使用边聚集系数(edge clustering coefficient,ECC) [ 16] 和皮尔逊相关系数来计算蛋白质关键性. 边聚集系数用于刻画网络中某个节点与其邻居节点的亲疏程度,给定PPI网络中的2个蛋白质节点 x和 y,用 N( x)、 N( y)分别表示节点 x和 y的直接邻居集合,则 x和 y的边聚集系数定义为
Ecc( x, y) =
Ecc( x, y)的取值范围是[0,1],其值越大,表明节点 x和 y联系越紧密 . 依据关键蛋白质与邻近蛋白质的相互作用较为紧密,且倾向于共表达的事实 [17 - 18] ,本文采用文献[19]的方法计算蛋白质节点 x的关键性度量 . 关键性度量的定义为
Pec( x) =
Pec( x)不但考虑了节点 x和 y在网络中的亲疏程度,而且增加了节点 x和 y的共表达强度对关键性度量的影响,因此能有效地评价一个蛋白质的关键性 [ 19] . 鉴于基因表达数据与PPI网络数据的差别,Pec( x)取值在不同PPI网络上有所偏差 . 为了易于比较,对给定网络中计算得到的关键性度量进行归一化,将其取值范围转化到[0,1],所得值就是给定PPI网络中蛋白质的关键性,归一化方法为
Est( x) =
式中max Pec和min Pec分别为整个网络中所有蛋白质关键性度量的最大值和最小值 . FACC-FMD在每次拾起放下之前会判断蛋白质的关键性是否大于阈值 θ,如果关键性小于 θ,不对它进行拾起放下操作,所以在蚁群聚类花费的时间肯定少于ACC-FMD .
FACC-FMD在蚁群聚类过程中,根据节点与核心组蛋白质的相似度计算拾起放下概率模型,再利用该模型完成聚类 . FACC-FMD的拾起概率模型和放下概率模型分别定义为
P p( i) =
P d( i) =
式中 s( i,Core)为蛋白质节点 i与核心组蛋白质的相似度 . 令 D j 为核心组蛋白质 j的度, D Core为核心组蛋白质的度之和,则 s( i,Core)由 i与每个核心组蛋白质的相似度进行加权求和得出,定义为
s( i,Core) =
FACC-FMD在蚁群聚类过程中用式(17) ~(19)替换公式(3) ~(5) .
2.4 算法描述与分析
FACC-FMD利用基因表达信息刻画蛋白质之间的共表达特性,再结合蛋白质在PPI网络中的拓扑特性,抽取核心组蛋白质,利用与核心组蛋白质的相似度计算拾起放下概率模型,依据概率模型进行拾起放下操作. 由于核心组蛋白质之间的差异性较大,初始聚类结果不需要合并过滤操作. 在聚类过程中依据关键性对拾起放下操作进行严格的约束,减少了拾起放下次数. 综上所述,相比ACC-FMD,FACC-FMD不需要进行合并过滤操作,且具有较少的拾起放下操作次数,因此会拥有较高的检测效率. 新算法的框架如算法1所示.
算法1:FACC-FMD
输入:PPI网络数据、基因表达信息、基因本体信息.
输出:功能模块集合 M 1, M 2,…, M k.
步骤1 初始化参数:蚂蚁数量 M、蚁群迭代数 T、拾起参数 k p、放下参数 k d、聚类系数阈值 ω、核心密度阈值 λ、核心共表达阈值 δ、关键性阈值 θ.
步骤2 计算节点 i的关键性Est:
for i = 1 to |V| do
compute Est( V i );
end for
步骤3 构建功能模块核心集合Core:
Core =⌀;
for i = 1 to |V| do
if( φ i ≥ ω) do
Core =Core∪ V i ;
end if
end for
步骤4 扩展功能模块核心,修正不符合阈值的功能模块核心:
for i = 1 to |Core | do
Core i= Core i ∪Neigh(Core i );
while
(Density(Core i ) <λ && coExp(Core i ) <δ)
do
Core i= Core i- min Est(Core i );
end for
步骤5 蚁群聚类过程
for t =1 to T do
for m=1 to M do
for i=1 to |Core | do
for V j ∈Neigh(Core i ) do
if Est( V i ) >θ do
利用拾起和放下规则对节点进行聚类;
end if
end for
蚂蚁 m得到自身的解;
end for
第 t代蚂蚁全部得到自身的解;
根据模块度评价标准,求出本次迭代的最优聚类结果,并利用聚类结果更新相似度;
end for
end for
从多次迭代中选取整个种群的最优解
步骤6 输出最优解 .
基于算法1的描述,对FACC-FMD算法做一个简单的分析:步骤1初始化参数的时间复杂度为常数级,即 O(1) . 步骤2计算PPI网络中每个节点的关键性 . 假设PPI网络中节点度的最大值为 d max,计算任意一个节点的关键性的时间复杂度为 O(
3 实验测试与分析
3.1 实验数据
本文使用通用的MIPS数据集 [ 20] 和DIP数据集 [ 21] 验证算法效果. 表1列出了数据集的详细信息. 实验中所需要的基因表达信息版本为GSE3431,包含7079个蛋白质的36个下的基因表达值,可通过网址http:∥www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE3431查询到. 为了评估检测结果的质量,使用文献[22]提供的标准数据集,该数据集包含428个功能模块. 实验环境是一台Windows 7 64位PC机,处理器型号是Intel i5-3470,3.2GHz CPU,4GB内存.
表 1 实验中所用到的数据集Table 1. Experimental data sets数据集 版本 网址 节点数 边数 DIPCore Scere20150429core http:∥dip.doe-mbi.ucla.edu/ 2452 5362 DIPFull Scere20150429full http:∥dip.doe-mbi.ucla.edu/ 5103 22817 MIPS PPI18052006 ftp:∥ftpmips.gsf.de/yeast/PPI/ 4545 12318 3.2 评价标准
本文选用2种流行的评价功能模块检测方法性能的度量标准 [ 21] .
3.2.1 精度、召回率、F度量
为了度量所预测的功能模块与标准功能模块之间的匹配度,大多数学者使用邻域亲和(NA)评分来进行计算:
NA( p, b) =
式中: p=( V p , V p )表示预测的功能模块; b=( V b , V b )表示标准的功能模块 . 若NA( p, b) >ω,则认为预测模块 p与标准模块 b相匹配(一般取 ω=0 .2或0 .25) . 本文实验中 ω=0 .2 .
精度(precision)、召回率(recall) 和F度量(F-measure)是PPI网络功能模块检测中常用的一组评价指标 . 令 P为算法预测的功能模块集合, B为标准功能模块集合,则 P中至少与一个标准功能模块相匹配的模块数量可表示为 N cp =|{ p|p∈ P,∃ b∈ B,NA( p, b)≥ ω} |;相对地, B中至少与一个预测的模块相匹配的模块数量为 N cb =|{ b|b∈ B,∃ p∈ P,NA( p, b)≥ ω} |. 于是,功能模块检测方法的精度和召回率的定义为
Precision =
Recall =
F-measure是精度和召回率的综合指标,用于衡量整体性能,其大小为精度和召回率的调和平均值,即
F-measure=
3 .2 .2 灵敏度、正的预测率、准确度
灵敏度(sensitivity,Sn)、正的预测率(positive predictive value,PPV)和准确度(accuracy,Acc)是最近提出的另一组评价模块检测方法性能的度量指标 . 设 m=|V p| , n=|V b| , T ij 为标准模块 p i 和预测模块 b j 共有的蛋白质数量,灵敏度和正的预测率定义为
Sn =
PPV =
式中: N i 为标准模块 i中所包含的蛋白质数量; T j=
Acc =
这6种评价指标都是以数值形式来度量检测质量,它们的取值范围都是[0,1],值越大,表示该项指标越好.
3.3 实验比较
为了取得更好的实验结果,对FACC-FMD算法中涉及的参数做了实验,每种参数在其他参数不变的情况下独立运行10次,以精度、召回率、F度量、灵敏度、正的预测率和准确度作为评估的指标,取10次运行的平均结果. 通过综合比较6种评估指标来选取参数. 经过实验比较,FACC-FMD算法参数设置如下:蚂蚁个数 M=50,最大种群迭代数 T=20,聚类系数阈值 ω=0.65(DIPFull和MIPS中 ω=0.35),拾起参数 k p =0.9,放下参数 k d =0.2,核心组蛋白质密度阈值 λ=0.7,核心组蛋白质共表达阈值 δ=0.15,蛋白质的关键性阈值 θ=0.3. 用于比较的其他算法参数尽量保持与原论文中一致.
3.3.1 与原始ACC-FMD算法比较
为了验证本文提出的2个策略的有效性,在DIPCore、DIPFull和MIPS三个数据集上分别做了不同的实验. 分别在ACC-FMD基础上单独使用核心策略、单独使用关键性策略和同时使用这2个策略(即FACC-FMD)进行了对比,并且每个实验均使用了相同的参数.
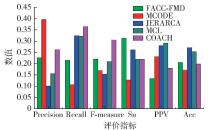
表2展示了2种策略对时间性能的影响. 从 表2可以看出,使用核心策略后,在3个数据集上的聚类时间分别增长到了1.7倍、1.4倍和1.8倍,这是因为聚类时需要计算蛋白质节点与整个核心组蛋白质的相似度,增大了蚂蚁拾起、放下模型的计算量;但是由于在进行后处理操作时,并未进行合并过滤操作,仅仅是计算了所有模块间的相似度,因此总的运行时间大幅降低,分别缩短了75.4%、94.4%和48.6%. 类似地,使用关键性策略后,在3个数据集上的聚类时间分别缩短了58.1%、68.0%和38.8%,这是因为关键性策略减少了拾起、放下的次数;总运行时间分别缩短了83.5%、96.1%和63.6%. 不难发现,同时使用核心策略和关键性策略即FACC-FMD,可以使得在3个数据集上的总运行时间分别缩短了86.4%、95.6%和76.9%. 因此,FACC-FMD的2个策略能够显著改善算法的时间性能.
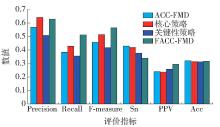
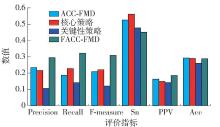
表 2 2种策略对运行时间的影响Table 2. Effects of two strategies on running time数据集 算法 聚类时间/s 合并过滤时间/s 总运行时间/s ACC-FMD 26.075 170.539 196.614 DIPCore 核心策略 45.334 3.025 48.359 关键性策略 10.932 21.533 32.465 FACC-FMD 26.719 26.719 ACC-FMD 469.699 17156.081 17625.780 DIPFull 核心策略 671.132 311.265 982.397 关键性策略 150.447 530.517 680.964 FACC-FMD 769.418 769.418 ACC-FMD 132.015 389.765 521.780 MIPS 核心策略 236.556 31.594 268.150 关键性策略 80.850 109.572 190.422 FACC-FMD 120.513 120.513 图3~5展示了2种策略对检测质量的影响,可以看到,单独使用核心策略能够使召回率和F度量得到提升,这是因为根据节点与整个核心组蛋白质的相似度进行聚类能够模拟蚁群聚类思想中一个数据与某块数据的相似度,使得算法具有更高的准确性. 单独使用关键性策略在精度、召回率和F度量上与ACC-FMD算法相比会有小幅下降,但由于关键性高的蛋白质拾起放下次数并未大幅减少,而关键性高的蛋白质对功能模块更重要,预测到的功能模块更接近标准模块,检测结果的正的预测率指标反而有所提升. 从这3张图能够看出,当2种策略结合时即FACC-FMD方法,除了灵敏度之外,检测质量整体要优于ACC-FMD. 灵敏度之所以有小幅下降,是因为FACC-FMD在聚类时,根据关键性对拾起放下做了更严格的约束,关键性过低的蛋白质节点在聚类中不参与拾起放下,而这些蛋白质也可能是标准功能模块内的蛋白质,导致对标准功能模块内的蛋白质覆盖率降低. 根据以上的分析,可以得知,与ACC-FMD相比,FACC-FMD得到了更具有竞争性的结果.
3.3.2 与其他经典算法比较
为了进一步展现FACC-FMD的整体性能,下面将该算法与MCODE、JERARCA、MCL和COACH四种经典的PPI网络功能模块检测方法进行了实验比较,5种算法在3个不同数据集上的实验结果如 表3~5所示.
对于每种算法, 表3~5列出了功能模块检测结果的模块数、模块平均大小、覆盖率、至少与一个实际模块相匹配的预测模块数( N cp)和至少与一个预测模块相匹配的实际模块数( N cb). 以DIPCore数据集为例,FACC-FMD算法检测到500个功能模块,其中有311个功能模块和216个标准模块相匹配. 检测到的每个功能模块大约含有9.06个蛋白质节点. 综合上面3个表格可以看出,对于 N cp指标,FACC-FMD算法在DIPCore、DIPFull和MIPS三个数据集上分别取得了第1、第1和第2的结果. 相对地,对于 N cb指标,FACC-FMD算法在DIPCore、DIPFull和MIPS三个数据集上分别取得了第3、第3和第4的结果. 这说明FACC-FMD检测结果中有多个功能模块匹配到相同的标准功能模块,所以检测到的标准功能模块种类较少. 下面用具体的评价标准对FACC-FMD与其他算法比较.
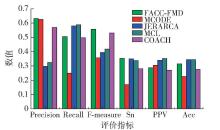
![]() 图 3 DIPCore数据集上2种策略检测质量的比较Figure 3. Comparative result of algorithms for DIPCore data
图 3 DIPCore数据集上2种策略检测质量的比较Figure 3. Comparative result of algorithms for DIPCore data![]() 图 4 DIPFull数据集上2种策略检测质量的比较Figure 4. Comparative result of algorithms for DIPFull data表 3 5种算法在DIPCore数据集的实验结果Table 3. Experimental results of five algorithms in DIPCore data sets
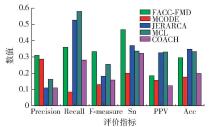
图 4 DIPFull数据集上2种策略检测质量的比较Figure 4. Comparative result of algorithms for DIPFull data表 3 5种算法在DIPCore数据集的实验结果Table 3. Experimental results of five algorithms in DIPCore data sets算法 模块数 模块平均大小 N cp N cb FACC-FMD 500 9.06 311 216 MCODE 107 5.52 67 107 JERARACA 160 4.57 248 266 MCL 527 4.65 171 252 COACH 350 7.33 199 213 表 4 5种算法在DIPFull数据集的实验结果Table 4. Experimental results of five algorithms in DIPFull data sets算法 模块数 模块平均大小 N cp N cb FACC-FMD 790 30.87 244 154 MCODE 70 13.11 20 36 JERARACA 1386 3.68 152 225 MCL 1204 4.24 196 248 COACH 1289 26.68 142 120 表 5 5种算法在MIPS数据集的实验结果Table 5. Experimental results of five algorithms in MIPS data sets算法 模块数 模块平均大小 N cp N cb FACC-FMD 507 45.17 115 92 MCODE 63 8.33 25 46 JERARACA 1012 4.49 102 139 MCL 593 6.16 92 138 COACH 1387 17.14 289 156 5种算法的2组指标的实验对比如 图6~8所示. 在DIPCore数据集上,
![]() 图 6 5种算法在DIPCore数据集上的比较结果Figure 6. Comparative result of five algorithms for DIPCore data
图 6 5种算法在DIPCore数据集上的比较结果Figure 6. Comparative result of five algorithms for DIPCore data![]() 图 7 5种算法在DIPFull数据集上的比较结果Figure 7. Comparative result of five algorithms for DIPFull data
图 7 5种算法在DIPFull数据集上的比较结果Figure 7. Comparative result of five algorithms for DIPFull dataFACC-FMD在精度、F度量和灵敏度指标上取得了最好的结果. 具体来说,FACC-FMD的精度是63.2%,比MCODE、JEARARCA、MCL和COACH分别高出0.6%、33.4%、30.8%和6.3%;F度量是55.7%,比MCODE、JEARARCA、MCL和COACH分别高出20.0%、16.4%、13.9%和2.6%;灵敏度是35.3%,比MCODE、JEARARCA、MCL和COACH分别高出18.4%、0.4%、1.5%和7.2%. 召回率、正的预测率和准确度分别位于第3、第4和第3. 在这3个指标上,JERARCA和MCL取得了最好的结果. 在DIPFull数据集上对比的结果类似,FACC-FMD的精度、F度量和灵敏度3个指标同样取得了最好的结果. 召回率、正的预测率和准确度都取得了第3的结果. 在MIPS数据集上,FACC-FMD的F度量取得了第2好的结果,灵敏度取得了最好的结果. 精度、召回率、正的预测率和准确度分别位于第3、第4、第5和第3. 通过这组实验可以看出,FACC-FMD在灵敏度这一指标上的表现很出色,说明FACC-FMD的检测结果对标准功能模块中的蛋白质具有较高的覆盖率. 这是因为FACC-FMD根据与核心组蛋白质的相似度进行聚类,也进一步证明了核心组蛋白质能够很好地标注一个功能模块. 同时,FACC-FMD在召回率上的表现不佳,造成该现象的主要原因是检测出的功能模块包含更多的蛋白质节点,使得 N cb的值较小. 由于MIPS网络噪声数据较多,影响了检测质量,因此正的预测率在5个算法中最差. 综合3个数据集的检测结果能够得出,FACC-FMD与一些经典算法相比在多项性能指标上都取得了较好的结果.
4 结论
1) 本文提出了一种快速的基于蚁群聚类的PPI网络功能模块检测方法FACC-FMD. 该方法结合基因表达信息刻画蛋白质之间的共表达特性,抽取核心组蛋白质,并计算蛋白质的关键性,依据关键性对拾起放下进行严格的约束,降低了聚类过程的计算复杂度,并且不需要对初始聚类结果进行合并和过滤操作,大大地提升了蚁群聚类算法在功能模块检测问题上的时间性能. 同时,检测质量也有所提升.
2) 在多个PPI网络上的实验表明,FACC-FMD和一些经典算法相比在多项性能指标上也具有一定优势.
The authors have declared that no competing interests exist. -
![]()
图 3 DIPCore数据集上2种策略检测质量的比较
Figure 3. Comparative result of algorithms for DIPCore data
![]()
图 4 DIPFull数据集上2种策略检测质量的比较
Figure 4. Comparative result of algorithms for DIPFull data
![]()
图 6 5种算法在DIPCore数据集上的比较结果
Figure 6. Comparative result of five algorithms for DIPCore data
![]()
图 7 5种算法在DIPFull数据集上的比较结果
Figure 7. Comparative result of five algorithms for DIPFull data
表 1 实验中所用到的数据集
Table 1 Experimental data sets
数据集 版本 网址 节点数 边数 DIPCore Scere20150429core http:∥dip.doe-mbi.ucla.edu/ 2452 5362 DIPFull Scere20150429full http:∥dip.doe-mbi.ucla.edu/ 5103 22817 MIPS PPI18052006 ftp:∥ftpmips.gsf.de/yeast/PPI/ 4545 12318  下载: 导出CSV
下载: 导出CSV
表 2 2种策略对运行时间的影响
Table 2 Effects of two strategies on running time
数据集 算法 聚类时间/s 合并过滤时间/s 总运行时间/s ACC-FMD 26.075 170.539 196.614 DIPCore 核心策略 45.334 3.025 48.359 关键性策略 10.932 21.533 32.465 FACC-FMD 26.719 26.719 ACC-FMD 469.699 17156.081 17625.780 DIPFull 核心策略 671.132 311.265 982.397 关键性策略 150.447 530.517 680.964 FACC-FMD 769.418 769.418 ACC-FMD 132.015 389.765 521.780 MIPS 核心策略 236.556 31.594 268.150 关键性策略 80.850 109.572 190.422 FACC-FMD 120.513 120.513
下载: 导出CSV
表 3 5种算法在DIPCore数据集的实验结果
Table 3 Experimental results of five algorithms in DIPCore data sets
算法 模块数 模块平均大小 N cp N cb FACC-FMD 500 9.06 311 216 MCODE 107 5.52 67 107 JERARACA 160 4.57 248 266 MCL 527 4.65 171 252 COACH 350 7.33 199 213
下载: 导出CSV
表 4 5种算法在DIPFull数据集的实验结果
Table 4 Experimental results of five algorithms in DIPFull data sets
算法 模块数 模块平均大小 N cp N cb FACC-FMD 790 30.87 244 154 MCODE 70 13.11 20 36 JERARACA 1386 3.68 152 225 MCL 1204 4.24 196 248 COACH 1289 26.68 142 120
下载: 导出CSV
表 5 5种算法在MIPS数据集的实验结果
Table 5 Experimental results of five algorithms in MIPS data sets
算法 模块数 模块平均大小 N cp N cb FACC-FMD 507 45.17 115 92 MCODE 63 8.33 25 46 JERARACA 1012 4.49 102 139 MCL 593 6.16 92 138 COACH 1387 17.14 289 156
下载: 导出CSV
-
[1] JI JZ,ZHANG AD,LIU CN,et al.Survey: functional module detection from protein-protein interaction networks[J].IEEE Transactions on Knowledge and Data Engineering,2014,26(2):261-277. [2] 冀俊忠,刘志军,刘红欣,等.蛋白质相互作用网络功能模块检测的研究综述[J].自动化学报,2014,40(4):577-593. JI JZ,LIU ZJ,LIU HX,et al.An overview of research on functional module detection for protein-protein interaction networks[J].Acta Automatica Sinica,2014,40(4):577-593. (in Chinese)
[3] 张媛,贾克斌,张爱冬.融合多数据源的蛋白质功能模块的挖掘算法[J].北京工业大学学报,2014,40(6):837-842. ZHANGY,JIA KB,ZHANG AD.Bipartite graph-based integrative method to detect consistent protein functional modules from multiple sources[J].Journal of Beijing University of Technology,2014,40(6):837-842. (in Chinese)
[4] BADER GD,HOGUE CW.An automated method for finding molecular complexes in large protein interaction networks[J].Bmc Bioinformatics,2003,4(1):1-27. [5] ALDECOAR,MARINI.Jerarca: efficient analysis of complex networks using hierarchical clustering[J].Plos One,2010,5(7):e11585. [6] VAN DONGENS.A cluster algorithm for graphs[J]. Report-Information Systems,2000(10):1-40. [7] MINW,LIX,KWOH CK,et al.A core-attachment based method to detect protein complexes in PPI networks[J].Bmc Bioinformatics,2009,10(11):169. [8] JI JZ,LIU ZJ,ZHANG AD,et al.Improved ant colony optimization for detecting functional modules in protein-protein interaction networks[C]∥Information Computing and Applications. Berlin: Springer,2012:404-413. [9] JI JZ,JIAOL,YANG CC,et al.MAE-FMD: multi-agent evolutionary method for functional module detection in protein-protein interaction networks[J].BMC bioinformatics,2014,15(1):325. [10] JI JZ,LIU HX,ZHANG AD,et al.ACC-FMD: ant colony clustering for functional module detection in protein-protein interaction networks[J].International Journal of Data Mining & Bioinformatics,2015,11(3):331-363. [11] LUMER ED,FAIETAB.Diversity and adaptation in populations of clustering ants[C]∥Proceedings of the Third International Conference on Simulation of Adaptive Behavior: From Animals to Animats 3.Brighton:MIT Press,1994:501-508. [12] TU BP,KUDLICKIA,ROWICKAM,et al.Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes[J].Science,2005,310(5751):1152-1158. [13] DEZSOZ,OLTVAI ZN,BARABASI AL.Bioinformatics analysis of experimentally determined protein complexes in the yeast Saccharomyces cerevisiae[J].Genome Research,2003,13(11):2450-2454. [14] WINZELER EA,SHOEMAKER DD,ASTROMOFFA,et al.Functional characterization of the Scerevisiae genome by gene deletion and parallel analysis[J].Science,1999,285(5429):901-906. [15] WATTSs DJ,STROGATZ SH.Collective dynamics of small-world networks[J].Nature,1998,393(6684):440-442. [16] PANGK,SHENGH,MAX.Understanding gene essentiality by finely characterizing hubs in the yeast protein interaction network[J].Biochemical & Biophysical Research Communications,2010,401(1):112-116. [17] ZHANG XX,XIAO QH,LIB,et al.Overlap maximum matching ratio (OMMR): a new measure to evaluate overlaps of essential modules[J].Frontiers of Information Technology & Electronic Engineering,2015,16(4):293-300. [18] LIM,LUY,WANGJ,et al.A topology potential-based method for identifying essential proteins from PPI networks[J].IEEE/ACM Transactions on Computational Biology & Bioinformatics,2015,12(2):372-383. [19] 李敏,张含会,费耀平.融合PPI和基因表达数据的关键蛋白质识别方法[J].中南大学学报(自然科学版),2013,44(3):1024-1029. LIM,ZHANG HH,FEI YP.Essential protein discovery method based on integration of PPI and gene expression data[J].Journal of Central South University (Science and Technology),2013,44(3):1024-1029. (in Chinese)
[20] PAGELP,KOVACS,OESTERHELDM,et al.The MIPS mammalian protein-protein interaction database[J].Bioinformatics,2005,21(6):832-834. [21] LIX,WUM,KWOH CK,et al.Computational approaches for detecting protein complexes from protein interaction networks: a survey[J].Bmc Genomics,2010,11(Suppl 1):1-19. [22] FRIEDEL CC,KRUMSIEKJ,ZIMMERR.Bootstrapping the interactome: unsupervised identification of protein complexes in yeast[J].Journal of Computational Biology,2009,16(8):971-987.
计量
- 文章访问数: 10
- HTML全文浏览量: 5
- PDF下载量: 5
